
はじめに:なぜ今、すべての製薬企業にPDE設定が求められるのか?
医薬品の品質、有効性、そして安全性を保証することは、製薬に関わるすべての企業にとって最も重要な使命です。近年、その安全性を確保する上で、グローバルな規制当局から強く求められているのが「PDE設定」です。本記事では、このPDE設定について、初めて担当される方や、改めて知識を整理したい品質保証・製造管理のご担当者様に向けて、その基本から具体的な算出プロセスまでを網羅的に解説します。
医薬品製造における「交叉汚染リスク」という見えざる脅威
複数の医薬品を同じ製造設備で製造する場合、ある医薬品の有効成分が、次に製造される別の医薬品に微量混入してしまう可能性があります。これが「交叉汚染(クロスコンタミネーション)」です。意図せず混入した有効成分は、患者様にとって予期せぬアレルギー反応や薬理作用を引き起こすリスクとなり得ます。この見えざる脅威を科学的根拠に基づいて管理することが、現代の医薬品製造における絶対的な要請となっています。
「PDEとは?」を分かりやすく解説:患者の安全を守るための科学的指標
PDE(Permitted Daily Exposure)は、日本語では「1日曝露許容量」と訳されます。これは、「ある物質を、生涯にわたって毎日体内に取り込んだとしても、健康への悪影響が予測されない量」を示す、物質固有の科学的な指標です。医薬品製造の文脈では、このPDE値を基準として、洗浄後に設備に残留しても安全と言える有効成分の量を算出します。つまりPDE設定は、感覚や慣習ではなく、毒性学的データに基づき患者様の安全を守るための、極めて重要なプロセスなのです。
本記事で得られること:PDE設定の全体像と規制を遵守するための実践的知識
この記事を最後までお読みいただくことで、以下の点を理解することができます。
・PDE設定がなぜ国際的な規制要件となったのか、その背景
・具体的なPDE算出のための5つのステップ
・複雑な計算式と、それを構成する「調整係数」の考え方
・PDE設定を成功させるために不可欠な専門家の役割
この知識は、貴社の品質保証体制をより強固にし、査察にも自信を持って対応できる体制を築くための一助となるはずです。
PDE設定の歴史的背景と規制要件
PDE設定がなぜこれほどまでに重要視されるようになったのでしょうか。その背景には、医薬品の品質管理に関する考え方の大きな変化と、国際的な規制の調和があります。
従来の「10ppm基準」「投与量0.1%基準」ではなぜ不十分なのか
かつて、洗浄バリデーションにおける残留限度値は、「次の製品への混入量を10ppm以下にする」「前製品の1日投与量の0.1%を基準とする」といった、一律の基準が便宜的に用いられてきました。しかし、これらの方法には大きな問題点がありました。それは、有効成分が持つ固有の「毒性の強さ」が全く考慮されていない点です。例えば、ごく微量でも強い薬理作用や毒性を示す成分と、比較的安全性の高い成分が、同じ基準で扱われてしまう可能性があったのです。
PIC/S GMPとHBELガイドラインがもたらしたグローバルスタンダードへの変革
このような状況を改善し、患者様の安全をより高いレベルで確保するため、PIC/S(医薬品査察協定及び医薬品査察共同スキーム)のGMPガイドラインでは、科学的根拠に基づく「健康ベース曝露限界値(HBEL: Health-Based Exposure Limit)」を用いて交叉汚染を管理することを義務化しました。PDEは、このHBELの最も代表的な指標です。この国際基準の導入により、毒性評価に基づいたリスク管理が、世界のデファクトスタンダードとなったのです。
査察で問われるポイント:科学的根拠に基づいた設定プロセスの重要性
国内外の規制当局によるGMP査察において、もはや単に洗浄バリデーションの記録があるだけでは十分ではありません。査察官が最も重視するのは、設定された残留限度値の「科学的妥当性」です。つまり、「なぜその値に設定したのか」という問いに対し、毒性学的データからPDE値を導き出すまでの論理的なプロセス全体を、明確に説明できることが求められます。
PDE設定は企業の責任:明確な方針確立の必要性
最終的に、自社で製造する医薬品のPDEを設定し、それに基づいてリスク管理を行う責任は、各製薬企業にあります。そのため、企業としてPDE設定に関する明確な方針と手順書を確立し、その方針に従って一貫性のある評価を実施していくことが不可欠です。これは、コンプライアンス遵守の観点だけでなく、企業の品質文化を示す上でも極めて重要と言えるでしょう。
【図解】専門家が教えるPDE算出の5つの重要ステップ
PDE設定は、複雑で専門性の高いプロセスですが、大きく5つのステップに分けることで、その全体像を理解しやすくなります。ここでは、各ステップで何を行うべきかを具体的に解説します。
ステップ1:ハザードの特定(情報収集と評価)
最初のステップは、評価対象となる有効成分の有害性(ハザード)に関する情報を、可能な限り網羅的に収集・評価することです。
どのような情報を収集すべきか?
収集すべき情報源は多岐にわたります。
■ 医薬品インタビューフォーム
■ CTD(コモン・テクニカル・ドキュメント)
■ 添付文書
■ 非臨床試験(動物実験)の報告書
■ 臨床試験(ヒトでの試験)の報告書
■ 学術論文や公的機関の評価書
これらの情報から、薬理作用、副作用、毒性に関するデータをすべて洗い出します。
収集した情報の信頼性評価とキースタディの選定
集めたすべてのデータが等しく重要というわけではありません。試験方法の妥当性や報告の質などを評価し、最も信頼性が高く、PDE設定の根幹(出発点)として用いるべき重要な試験(キースタディ)を選定します。
ステップ2:重大な影響の特定(クリティカルエフェクトの選択)
次に、収集したデータの中から、健康への影響を評価する上で最も重要となる作用(クリティカルエフェクト)を特定します。
最も感度の高い指標をどう見極めるか
クリティカルエフェクトとは、最も低い用量で認められる有害な影響のことです。これには、意図した薬理作用そのものや、臨床でみられる副作用、動物実験でのみ確認された毒性など、あらゆる影響が含まれます。この中から、最も敏感な指標を見つけ出すことが重要です。
可逆的影響と不可逆的影響の重要度
影響の性質も考慮されます。例えば、投与を中止すれば回復する可逆的な影響(例:一時的な肝機能数値の上昇)よりも、回復しない、あるいは極めて深刻な不可逆的な影響(例:発がん性、生殖発生毒性、神経毒性など)は、より重大な影響として扱われます。
ステップ3:POD(Point of Departure)の選定(NOAELの決定)
クリティカルエフェクトを特定したら、次はその影響が認められなかった最大用量を決定します。これがPDE算出の計算の出発点(POD: Point of Departure)となります。
NOAEL(無毒性量)とは何か?
PODとして最も一般的に用いられるのが、NOAEL(No-Observed-Adverse-Effect Level:無毒性量)です。これは、動物実験などにおいて、有害な影響が統計学的に認められなかった最大の投与量を指します(単位は通常 mg/kg/day)。
ヒトデータと動物データのどちらを優先すべきか
原則として、信頼できるヒトの臨床データが存在する場合は、それを最優先で用います。しかし、多くの場合は十分なヒトデータが存在しないため、動物実験のデータからNOAELを選定することが一般的です。
ステップ4:5つの調整係数(不確実性係数)の適用
動物実験で得られたNOAELを、そのままヒトの安全な量として用いることはできません。様々な不確実な要素を考慮し、安全側に立った評価を行うために、「調整係数(または不確実性係数)」を用いてNOAELを割り算し、より安全な値を導き出します。この係数の設定には、毒性学の深い専門知識が求められます。
| 係数 | 名称 | 内容 |
| F1 | 種差 | 動物実験の結果をヒトに当てはめる(外挿する)際の不確実性を考慮する係数。 |
| F2 | 個体差 | ヒト集団の中での感受性の違い(例:子供と大人、健康な人と病人)を考慮する係数。 |
| F3 | 曝露期間 | 短期間の毒性試験の結果から、生涯にわたる曝露の影響を推定する際の不確実性を考慮する係数。 |
| F4 | 影響の重篤性 | クリティカルエフェクトが特に重篤(例:催奇形性)である場合に、追加の安全性を確保するための係数。 |
| F5 | NOAELへの調整 | 明確なNOAELが得られず、最小の用量で影響が見られた値(LOAEL)しか無い場合などに用いる係数。 |
ステップ5:最終報告書(モノグラフ)の作成と文書管理
最後のステップは、これらステップ1から4までの全プロセスを、一つの報告書(モノグラフ)として詳細に文書化することです。
モノグラフに記載すべき必須項目とは
モノグラフには、収集した全データのリスト、クリティカルエフェクトとPODの選定理由、適用した各調整係数の妥当性、そして最終的なPDE値の算出過程を、第三者が読んでも完全に再現・検証できるように記載します。
製造委託先に提出する際の注意点
このモノグラフは、自社での保管はもちろん、製造を外部に委託(CMO/CDMO)する際には、委託先に提供する必要がある極めて重要な文書です。これにより、委託先も科学的根拠に基づいた適切な洗浄バリデーションを実施することが可能となります。
PDE算出式の徹底解説
ここでは、5つのステップを経て導き出された各要素を、どのように一本の式に落とし込み、最終的なPDE値を算出するのかを解説します。
PDE算出の基本式とその構成要素
PDEを算出するための国際的に認められた基本式は以下の通りです。
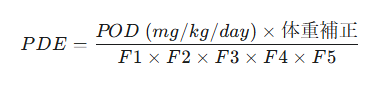
■ POD (Point of Departure): ステップ3で選定したNOAELなどの値(単位:mg/kg/day)。
■ 体重補正: ヒトの標準的な体重(通常は50kg)を乗じ、動物の体重あたりの投与量からヒト一人あたりの量に換算します。
■ F1~F5: ステップ4で科学的妥当性をもって設定した5つの調整係数。これらの積でPODを割り、安全性を確保します。
計算例で学ぶ:具体的な数値の当てはめ方(仮想ケーススタディ)
数式だけでは分かりにくいかもしれませんので、具体的な仮想ケースで計算してみましょう。
ケース:十分な動物データがある場合
前提条件:
・有効成分Aのラットを用いた90日間反復投与毒性試験のデータがある。
・クリティカルエフェクトは肝機能への影響と判断された。
・この影響が見られなかった最大の投与量(NOAEL)は 10 mg/kg/day であった。
・ヒトへの外挿(F1=5)、個体差(F2=10)、短期試験からの外挿(F3=5)を考慮し、その他の係数(F4, F5)は1と判断した。
計算プロセス:
1. PODとしてNOAELの 10 mg/kg/day を採用します。
2. ヒトの標準体重 50 kg を乗じます。
3. 調整係数の積は 5 (F1) × 10 (F2) × 3 (F3) = 150 となります。
4. これらを式に当てはめます。
PDE = (10 mg/kg/day × 50 kg) ÷ (5 × 10 × 3) = 3.3 mg/day ≒ 3 mg/day
この計算により、有効成分AのPDE値は 3 mg/day と算出されました。これは、毎日3mgまでであれば、生涯にわたり摂取し続けても健康への悪影響はないと科学的に推定される量、ということになります。
PDE設定における重要概念と関連用語
PDE設定のプロセスをより深く理解するために、関連する重要な概念や用語についても知っておくことが役立ちます。
PDEとOEL(職業曝露限界)の違いとは?
PDEと共によく耳にする言葉に「OEL(Occupational Exposure Limit:職業曝露限界)」があります。この二つは密接に関連していますが、その目的と対象が明確に異なります。
目的の違い:患者の安全 vs 作業者の安全
■ PDE: 交叉汚染を通じて医薬品を服用する「患者様」の安全を守るための指標です。曝露経路は主に経口や注射など、医薬品の投与経路が考慮されます。
■ OEL: 製造工場などで有効成分を取り扱う「作業者」の健康を守るための指標です。曝露経路は主に吸入(空気を吸い込むこと)が考慮され、空気1立方メートルあたりの濃度(例: μg/m³)で示されます。いわゆる「OEL設定」は、この作業環境の安全基準を定めるプロセスです。
算出アプローチの連携:PDEからOELの関係
PDEは主に「1日あたりに体内へ取り込まれても安全と考えられる量(mg/day)」を示すものであり、OELは「空気中濃度としての安全限界(mg/m³)」を示します。
この2つは直接的に換算できるものではありませんが、OEL設定の際にはPDEの情報を参考にできる場合があります。
例えば、体重50 kg、呼吸量10 m³/日を仮定した場合、理論上の目安として次のようにPDEをOELに換算することが考えられます:
OEL ≒ PDE ÷ 呼吸量 (※参考換算に過ぎません)
このように、PDE設定で得られた「全身許容量」の情報を基礎にして、作業環境の安全濃度(OEL)を検討することができます。
また、健康な成人作業者を対象とする場合、個体差の係数(F₂)を5とすることが一般的です。
産業衛生の視点:医薬品を「不純物」として捉える新しい考え方
PDE設定が従来の医薬品安全性評価と大きく異なるのは、その視点です。通常の安全性評価では、医薬品を「有効成分」として、その効果と副作用のバランスを評価します。一方、PDE設定では、交叉汚染の文脈において、ある医薬品の有効成分を、次に製造される製品にとっては「不純物」として捉えます。この産業衛生学的な視点に立つことで、患者様にとって意図しない曝露のリスクを客観的に評価することが可能になるのです。
なぜ「毒性学の専門家」による評価が不可欠なのか
これまで見てきたように、PDE設定は単一の正解がある単純な計算ではありません。
高度な専門知識と科学的判断が求められる理由
どの文献をキースタディとして採用するか、どの影響をクリティカルエフェクトと見なすか、そして特に5つの調整係数をいくつに設定するか。これらのプロセスには、毒性学、薬理学、統計学など、多岐にわたる分野の高度な専門知識と、豊富な経験に裏打ちされた科学的な「判断」が不可欠です。この判断の質が、最終的なPDE値の信頼性を大きく左右します。
まとめ:科学的根拠に基づくリスク管理で未来の安全を築く
本記事では、医薬品製造における交叉汚染リスク管理の核心である「PDE設定」について、その規制上の背景から、具体的な5つの算出ステップ、そして関連する重要概念までを包括的に解説しました。
PDE設定プロセスの重要ポイントの再確認
PDE設定は、もはや避けては通れないグローバルスタンダードです。その成功の鍵は、信頼できる情報の網羅的な収集、クリティカルエフェクトの適切な特定、そして科学的根拠に基づく調整係数の適用にあります。この一連のプロセスを、専門家の知見を活用しながら、透明性の高い文書として記録に残すことが極めて重要です。
貴社のコンプライアンスと製品の安全性を確実にするために
科学的根拠に基づいたPDE設定を社内方針として確立し、実践することは、規制当局の査察に対応するコンプライアンスの観点だけでなく、何よりも患者様へ安全な医薬品を届けるという、製薬企業の社会的責務を果たす上で不可欠です。堅牢なリスク管理体制は、企業の信頼性を高め、未来の成長を支える礎となります。
PDE設定に関するご相談はこちら
PDE設定のプロセスは複雑であり、多くの専門的判断を要します。「自社だけでの評価に不安がある」「第三者の客観的な評価が欲しい」など、お困りのことがございましたら、ぜひ一度、専門家にご相談ください。
医薬品の交叉汚染リスク管理に不可欠な「PDE設定」、そして労働安全衛生に関わる「OEL設定」に関するお悩みは、株式会社ケミカル・リスクアセス・ソリューションズまでご相談ください。科学的根拠に基づく信頼性の高い評価で、貴社の課題解決をサポートいたします。